Mölnlycke is MDR-certified
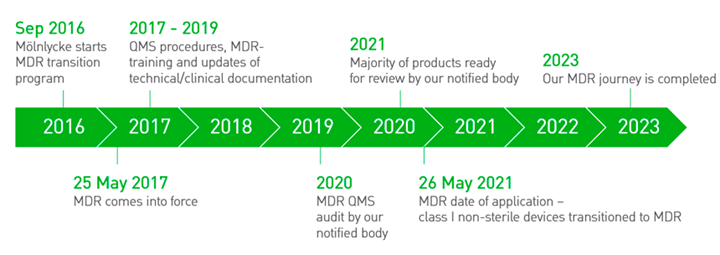
On 25 May 2017, The European Medical Device Regulation, ´MDR´, came into force, replacing the Medical Device Directive, ´MDD´. To remain on the market, class I non-sterile devices were required to be MDR certified by 26 May 2021, the MDR date of application. MDD-certificated products of other medical device classes will transition to MDR prior to the expiry of the validity of the respective MDD certificates.
Mölnlycke has been well prepared for the MDR transition. The internal MDR transition programme was initiated in September 2016, working to secure compliance with the MDR-obligations until Mölnlycke-manufactured products om the European market were MDR-certified. This journey was completed in June 2023.
Mölnlycke MDR timeline
The main three objectives for the new regulation
- Improve the health and safety level
- Continue to enhance the free trade of medical devices in EU
- Ensure products are current to state of art technology and scientific knowledge
The regulation was due to go live in May 2020 but was postponed by one year and came into force on May 26, 2021.
A directive can be interpreted different in each member state, a regulation is much more comprehensive. There are enhanced requirements on the safety and reliability of medical devices, with more emphasis on a life cycle approach, backed by clinical data, as well as increased post-market surveillance. National Competent Authorities and the EU Commission will have a stronger control and monitoring role. There are also added requirements that will strengthen transparency and information for users.
The outcome in the long run will be a stronger, more accountable MedTech industry that favours responsible businesses.
The purpose of the new regulation, as well as for all other regulations, is to create rules that will keep everyone safe, the people working with the products as well as the patients using them. The new regulation will help the clinicians to feel safe, by knowing that all products on the market under MDR certification have been through a tough assessment and have a high level of clinical evidence.
The main three objectives for the new regulation
- Improve the health and safety level
- Continue to enhance the free trade of medical devices in EU
- Ensure products are current to state of art technology and scientific knowledge
The regulation was due to go live in May this year but has been postponed one year and will go live on May 26, 2021. The absolute last day tosell products placed on the market under MDD certification is May 2025.
What are the main differences between MDD and MDR?
A directive can be interpreted different in each member state, a regulation is much more comprehensive. There are enhanced requirements on the safety and reliability of medical devices, with more emphasis on a life cycle approach, backed by clinical data, as well as increased post-market surveillance. National Competent Authorities and the EU Commission will have a stronger control and monitoring role. There are also added requirements that will strengthen transparency and information for users.
The outcome in the long run will be a stronger, more accountable MedTech industry that favours responsible businesses.
How has MDR impacted the industry?
MDR will affect all medical devices sold in EU, either they are manufactured in EU or imported. All devices, including legacy products, need to be recertified and get new EC-certification. Companies are also required to upgrade and recertify their quality management systems to meet the new regulations.
It is costly to implement and requires every medical device to have upgraded technical documentation. As a result, there are predictions that many existing products will not remain on the market.
At the beginning of April, while Mölnlycke were in a strong position, there was considerable concern that the introduction of MDR would aggravate the shortages of personal protective equipment (PPE) and other medical devices needed to fight coronavirus. The EU parliament has now agreed to postpone the MDR deadline by a year to allow authorities and the industry to prioritise their efforts towards COVID-19.
The postponement was a relief for many companies not as far down the line as Mölnlycke is, and more companies will now be able to meet the requirements on time.
What can our customers expect from the new regulation?
The purpose of the new regulation, as well as for all other regulations, is to create rules that will keep everyone safe, the people working with the products as well as the patients using them. The new regulation will help the clinicians to feel safe, by knowing that all products on the market after MDR is live have been through a tough assessment and have a high level of clinical evidence.
How does Mölnlycke meet the MDR timeplan?
The delay of MDR have not impacted the way Mölnlycke work to meet the new regulation. We were an early adopter of MDR and have already got our first issue of our MDR certificates which mean that we can already now start to CE mark some of our products in line with the new regulation.
We are well prepared and our timeplan still follows the original MDR dates. Our customers can be sure that we will continue to provide products with a high level of health and safety protection also after the new regulation is implemented.





